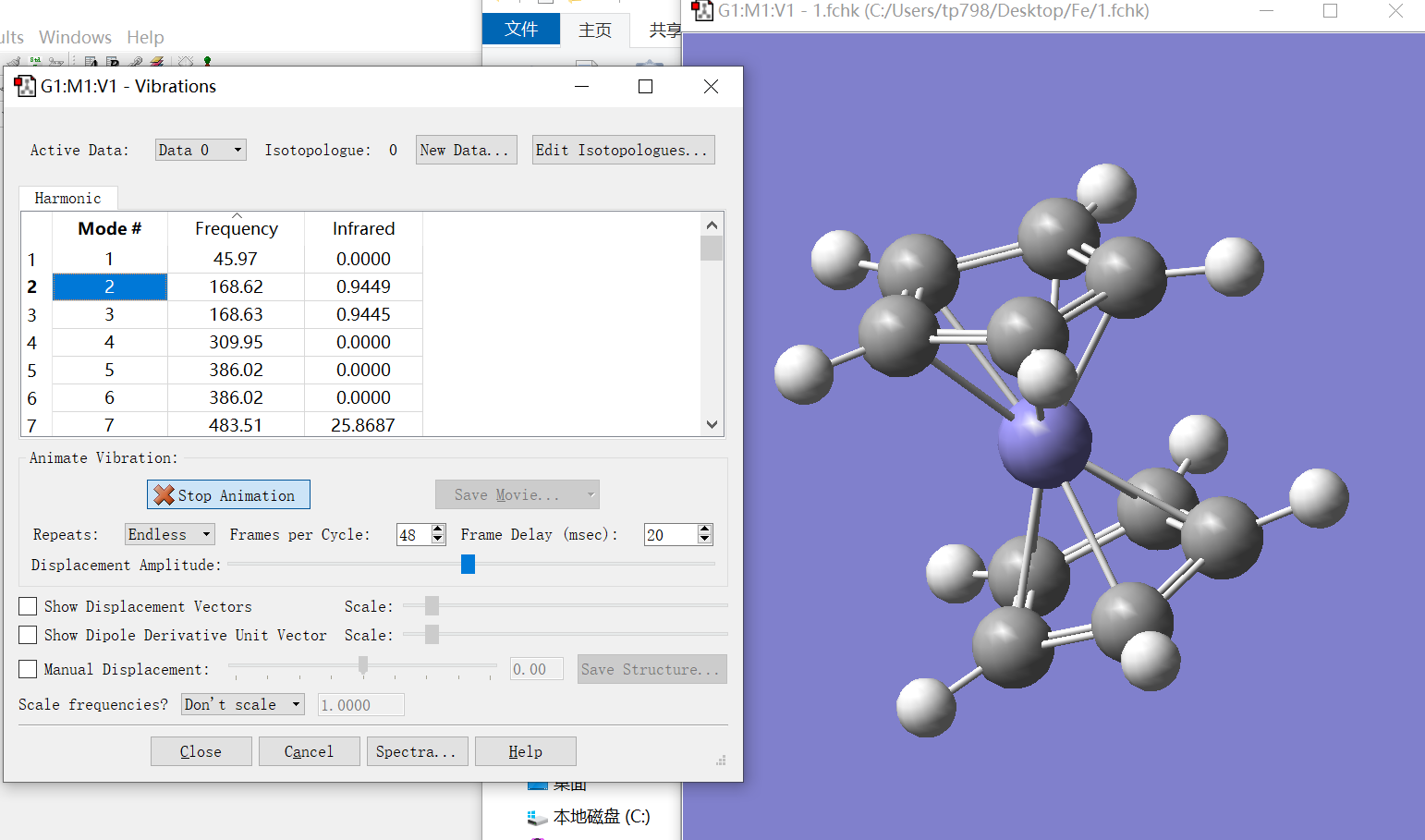
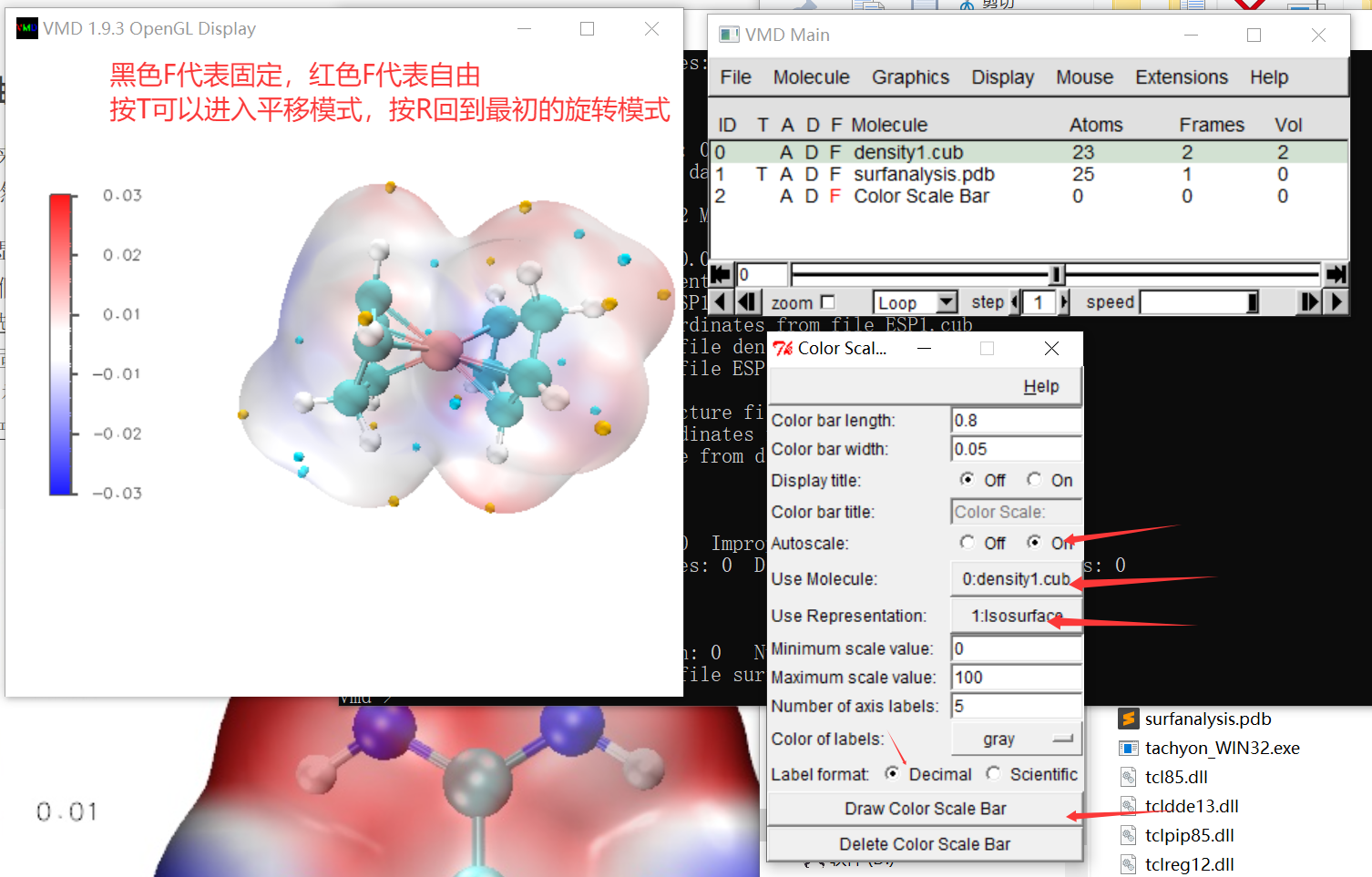
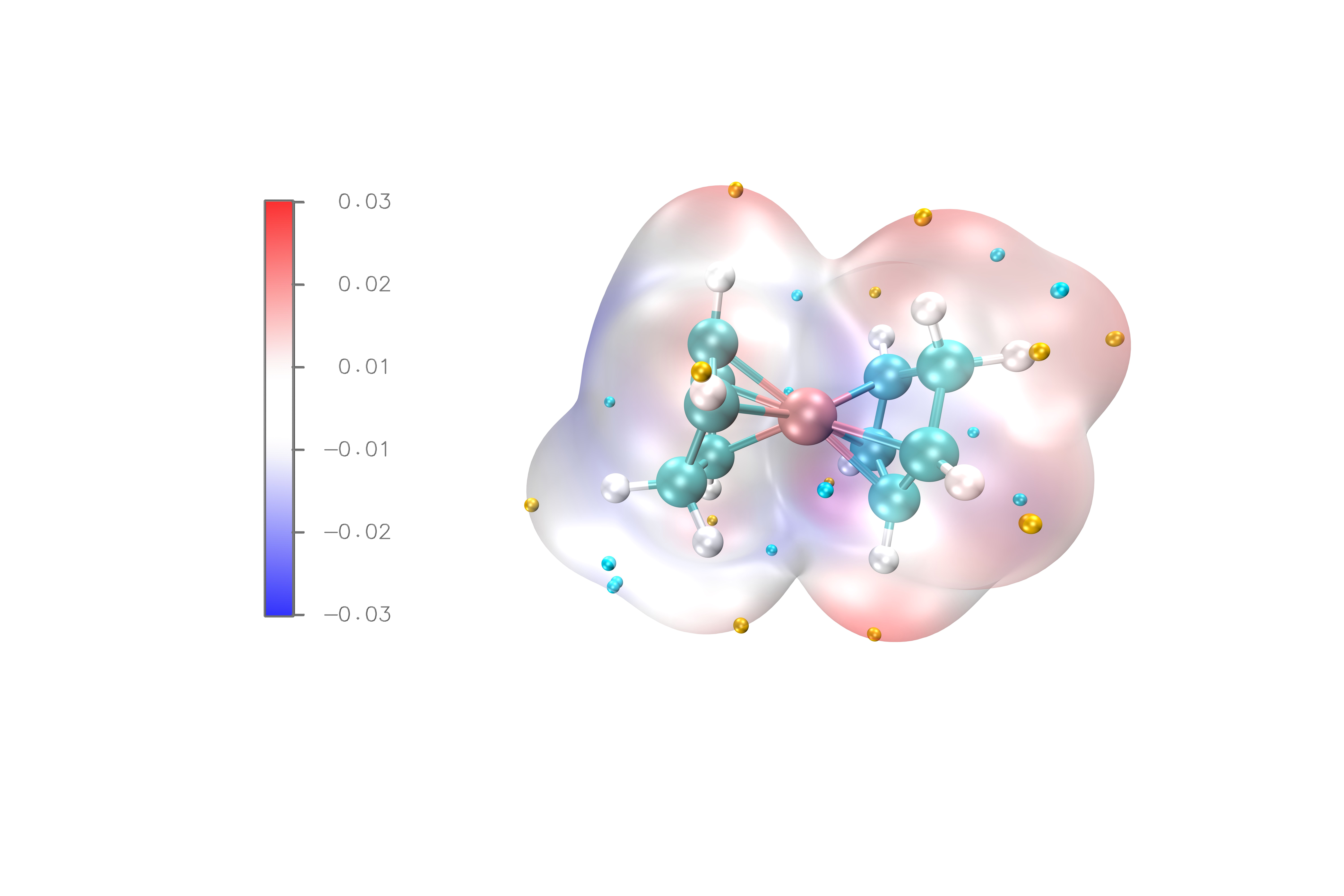
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
241
242
243
244
245
246
247
248
249
250
251
252
253
254
255
256
257
258
259
260
261
262
263
264
265
266
267
268
269
270
271
272
273
274
275
276
277
278
279
280
281
282
283
284
285
286
287
288
289
290
291
292
293
294
295
296
297
298
299
300
301
302
303
304
305
306
307
308
309
310
311
312
313
314
315
316
317
318
319
320
321
322
323
324
325
326
327
328
329
330
331
332
333
334
335
336
337
338
339
340
341
342
343
344
345
346
347
348
349
350
351
352
353
354
355
356
357
358
359
360
361
362
363
364
365
366
367
368
369
370
371
372
373
374
375
376
377
378
379
380
381
382
383
384
385
386
387
388
389
390
391
392
393
394
395
396
397
398
399
400
401
402
403
404
405
406
407
408
409
410
411
412
413
414
415
416
417
418
419
420
421
422
423
424
425
426
427
428
429
430
431
432
433
434
435
436
437
438
439
440
441
442
443
444
445
446
447
448
449
450
451
| PS C:\Multiwfn_3.8_dev_bin_Win64> ls
Directory: C:\Multiwfn_3.8_dev_bin_Win64
Mode LastWriteTime Length Name
---- ------------- ------ ----
d---- 2025/5/15 12:20 examples
-a--- 2025/5/23 9:20 8114156 1.fch
-a--- 2024/9/21 17:46 3057 使用Multiwfn发表文章必须在正文里进行引用(包括代算).txt
-a--- 2025/5/28 17:51 105 ESPext.bat
-a--- 2018/10/7 5:24 32 ESPext.txt
-a--- 2025/5/28 17:51 460 ESPiso.bat
-a--- 2021/11/18 10:43 26 ESPiso.txt
-a--- 2025/5/28 17:51 344 ESPpt.bat
-a--- 2018/9/30 13:53 29 ESPpt.txt
-a--- 2025/2/9 9:28 200205 How to cite Multiwfn.pdf
-a--- 2023/3/2 2:26 2047000 libiomp5md.dll
-a--- 2024/8/27 20:00 1254 LICENSE.txt
-a--- 2025/4/13 9:17 416416 Multiwfn quick start.pdf
-a--- 2025/5/5 2:50 35666432 Multiwfn.exe
-a--- 2025/5/16 1:26 20490 settings.ini
PS C:\Multiwfn_3.8_dev_bin_Win64> .\ESPiso.bat
C:\Multiwfn_3.8_dev_bin_Win64>Multiwfn 1.fch -ESPrhoiso 0.001 0<ESPiso.txt
Multiwfn -- A Multifunctional Wavefunction Analyzer
Version 3.8(dev), update date: 2025-May-5
Developer: Tian Lu (Beijing Kein Research Center for Natural Sciences)
Multiwfn official website: http:
Multiwfn English forum: http:
Multiwfn Chinese forum: http:
( Number of parallel threads: 4 Current date: 2025-05-30 Time: 20:33:12 )
Both following papers ***MUST BE CITED IN MAIN TEXT*** if Multiwfn is used:
Tian Lu, Feiwu Chen, J. Comput. Chem., 33, 580 (2012) DOI: 10.1002/jcc.22885
Tian Lu, J. Chem. Phys., 161, 082503 (2024) DOI: 10.1063/5.0216272
See "How to cite Multiwfn.pdf" in Multiwfn binary package for more information
Please wait...
Loading various information of the wavefunction
The highest angular moment basis functions is F
Loading basis set definition...
Loading
Converting
Back
Generating
Generating
Total/Alpha/Beta
Net
Atoms: 23, Basis
Total
This
Orbitals
Title
Loaded
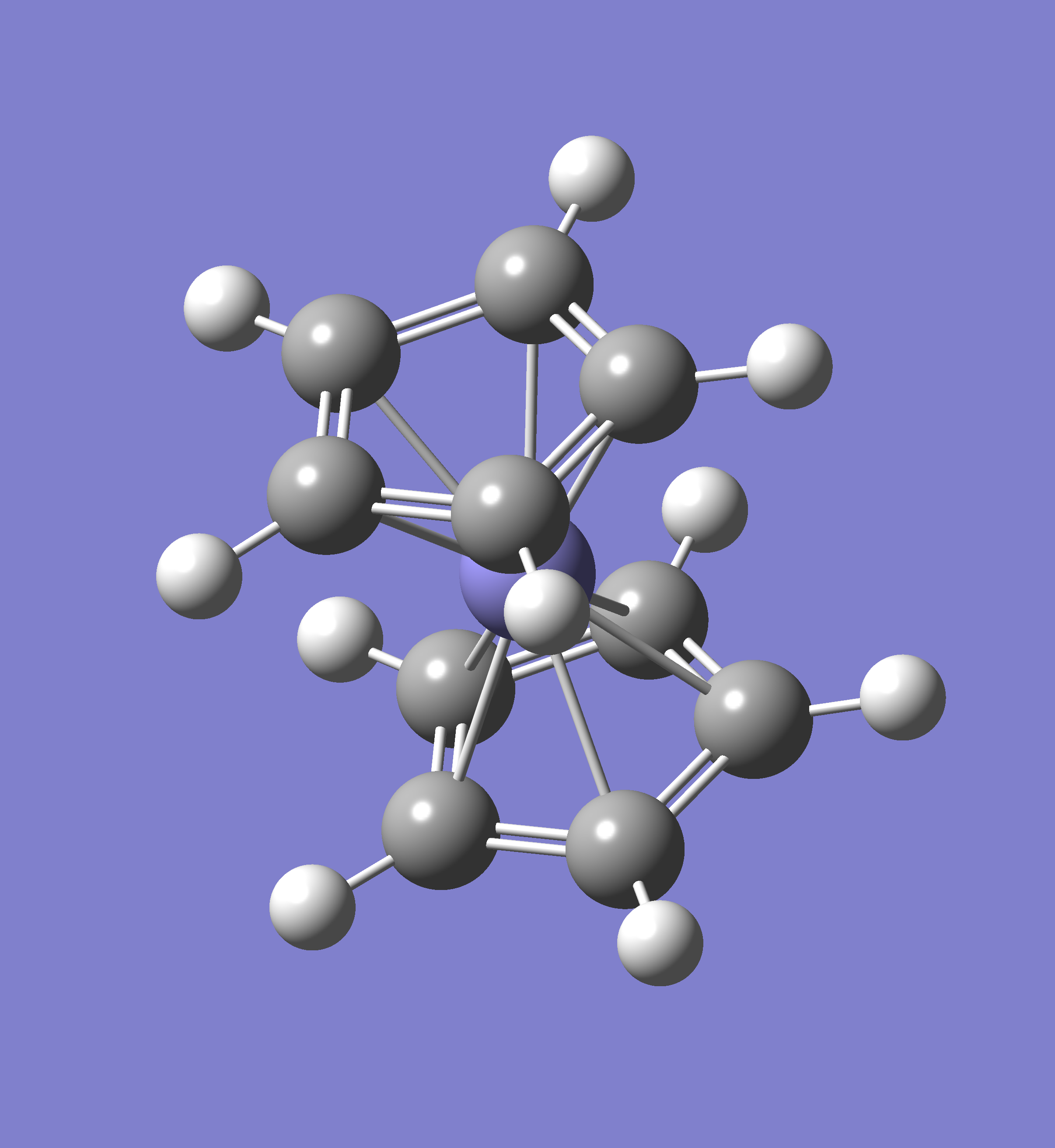
Formula: H12
Molecule
Point
"q": Exit
************ Main
0 Show
1 Output
3 Output
4 Output
5 Output
6 Check
7 Population
8 Orbital
10 Plot
11 Plot /Raman//ECD//ROA/
12 Quantitative
13 Process
14 Adaptive
15 Fuzzy
16 Charge
17 Basin
19 Orbital
21 Energy
23 ETS-NOCV
25 Electron
26 Structure
100 Other
300 Other
-10 Return
-2 Obtain
-1 Obtain
0 Set
----------- Available
1 Electron
4 Value
5 Electron
6 Hamiltonian
7 Lagrangian
8 Electrostatic
9 Electron
10 Localized
11 Local
12 Total
13 Reduced
15 Sign(lambda2)*rho
17 Correlation
18 Average
19 Source
20 Electron ;d) 21 Orbital overlap dist. func. D(r)
22 Delta-g (promolecular approximation) 23 Delta-g (Hirshfeld partition)
24 Interaction region indicator (IRI) 25 van der Waals potential (probe=C )
100 User-defined function (iuserfunc= -1), see Section 2.7 of manual
Please select a method to set up
-10 Set ~
1 Low
2 Medium
3 High
4 Input
5 Input
6 Input
7 The
8 Use
10 Set
11 Select
100 Load
Coordinate
Coordinate
Grid
Number
Note: Virtual
Note: All
Unique
Progress: [##################################################] 100.0 % \
Calculation
Note: Previous
Electric
X
Y
Z
Total
The
The
Summing
100.725159050150
Summing
100.725159050150
Summing
0.000000000000000E+000
---------- Post-processing
-1 Show
0 Return
1 Save
2 Export
3 Export
4 Set
5 Multiply
6 Divide
7 Add
8 Substract
9 Multiply
Exporting
Done! Grid
Hint: If
---------- Post-processing
-1 Show
0 Return
1 Save
2 Export
3 Export
4 Set
5 Multiply
6 Divide
7 Add
8 Substract
9 Multiply
Note: A
"q": Exit
************ Main
0 Show
1 Output
3 Output
4 Output
5 Output
6 Check
7 Population
8 Orbital
10 Plot
11 Plot /Raman//ECD//ROA/
12 Quantitative
13 Process
14 Adaptive
15 Fuzzy
16 Charge
17 Basin
19 Orbital
21 Energy
23 ETS-NOCV
25 Electron
26 Structure
100 Other
300 Other
-10 Return
-2 Obtain
-1 Obtain
0 Set
----------- Available
1 Electron
4 Value
5 Electron
6 Hamiltonian
7 Lagrangian
8 Electrostatic
9 Electron
10 Localized
11 Local
12 Total
13 Reduced
15 Sign(lambda2)*rho
17 Correlation
18 Average
19 Source
20 Electron ;d) 21 Orbital overlap dist. func. D(r)
22 Delta-g (promolecular approximation) 23 Delta-g (Hirshfeld partition)
24 Interaction region indicator (IRI) 25 van der Waals potential (probe=C )
100 User-defined function (iuserfunc= -1), see Section 2.7 of manual
Please select a method to set up
-10 Set ~
1 Low
2 Medium
3 High
4 Input
5 Input
6 Input
7 The
8 Use
10 Set
11 Select
100 Load
Coordinate
Coordinate
Grid
Number
Note: Virtual
Unique
Note: ESP
Detecting
Number
Initializing
LIBRETA
NOTE: The
Progress: [##################################################] 100.0 % \
Setting
Calculation
Note: Previous
The
The
Summing
3.07509251072690
Summing
8.67387639395045
Summing
-5.59878388322356
---------- Post-processing
-1 Show
0 Return
1 Save
2 Export
3 Export
4 Set
5 Multiply
6 Divide
7 Add
8 Substract
9 Multiply
Exporting
Done! Grid
Hint: If
---------- Post-processing
-1 Show
0 Return
1 Save
2 Export
3 Export
4 Set
5 Multiply
6 Divide
7 Add
8 Substract
9 Multiply
forrtl: severe
Image
Multiwfn.exe
Multiwfn.exe
Multiwfn.exe
Multiwfn.exe
Multiwfn.exe
KERNEL32.DLL
ntdll.dll
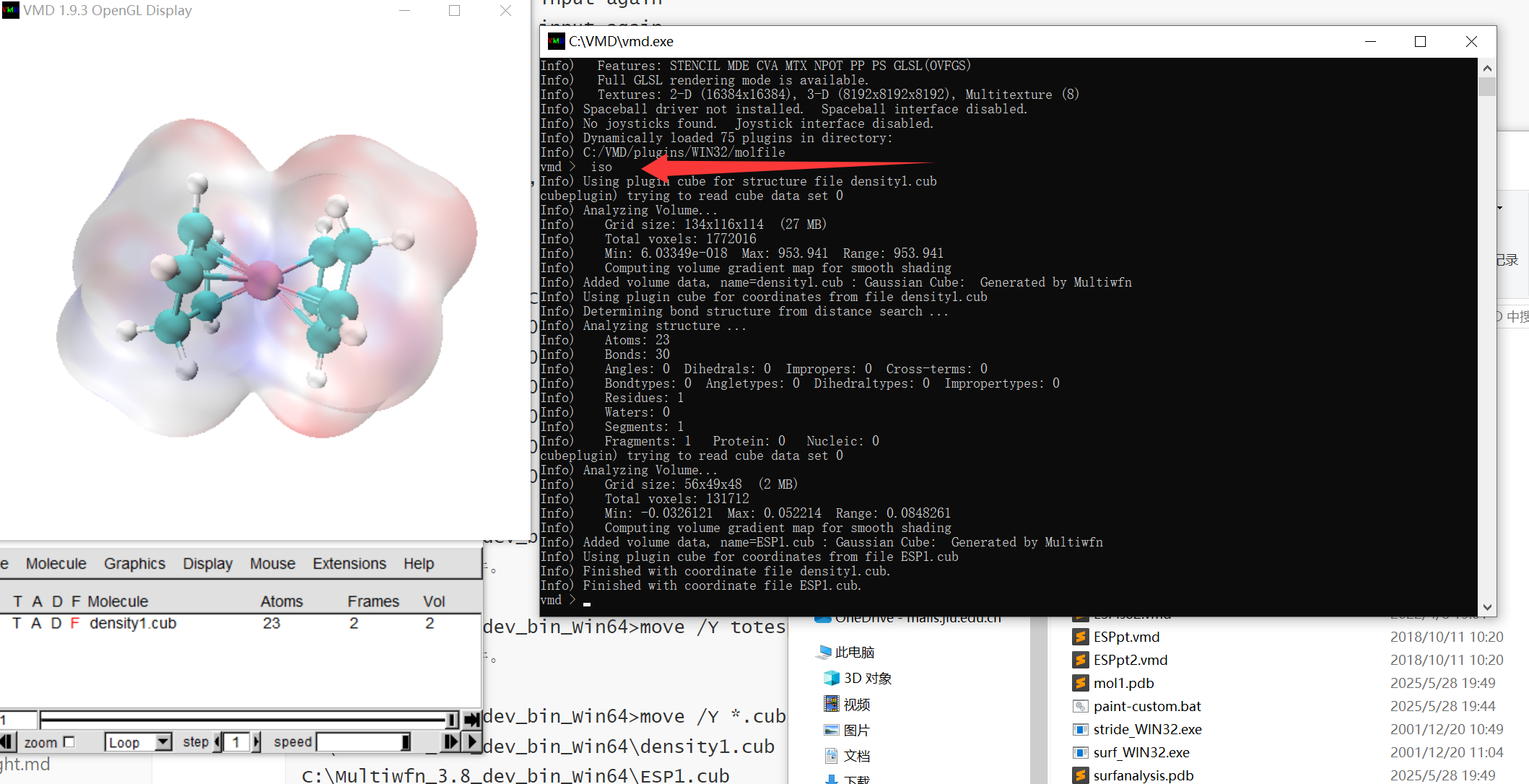
C:\Multiwfn_3.8_dev_bin_Win64>move /Y density.cub density1.cub
移动了 1 个文件。
C:\Multiwfn_3.8_dev_bin_Win64>move /
移动了 1 个文件。
C:\Multiwfn_3.8_dev_bin_Win64>Multiwfn
Error: Unable
Multiwfn
Version
Developer: Tian
Multiwfn ///multiwfn
Multiwfn English forum: http:
Multiwfn Chinese forum: http:
( Number of parallel threads: 4 Current date: 2025-05-30 Time: 20:33:30 )
Both following papers ***MUST BE CITED IN MAIN TEXT*** if Multiwfn is used:
Tian Lu, Feiwu Chen, J. Comput. Chem., 33, 580 (2012) DOI: 10.1002/
Tian /5.0216272
See in Multiwfn binary package for more information
Now input file path, for example, E:\Planetarian\Yumemi_Hoshino.mwfn
(.wfn//wfx//molden//xyz//cif/
Hint: Pressing
"5" cannot
"1" cannot
"3" cannot
"2" cannot
"0" cannot
"5" cannot
"12" cannot
"1" cannot
"2" cannot
forrtl: severe
Image
Multiwfn.exe
Multiwfn.exe
Multiwfn.exe
Multiwfn.exe
KERNEL32.DLL
ntdll.dll
C:\Multiwfn_3.8_dev_bin_Win64>move /Y density.cub density2.cub
系统找不到指定的文件。
C:\Multiwfn_3.8_dev_bin_Win64>move /
系统找不到指定的文件。
C:\Multiwfn_3.8_dev_bin_Win64>Multiwfn
Error: Unable
Multiwfn
Version
Developer: Tian
Multiwfn ///multiwfn
Multiwfn English forum: http:
Multiwfn Chinese forum: http:
( Number of parallel threads: 4 Current date: 2025-05-30 Time: 20:33:30 )
Both following papers ***MUST BE CITED IN MAIN TEXT*** if Multiwfn is used:
Tian Lu, Feiwu Chen, J. Comput. Chem., 33, 580 (2012) DOI: 10.1002/
Tian /5.0216272
See in Multiwfn binary package for more information
Now input file path, for example, E:\Planetarian\Yumemi_Hoshino.mwfn
(.wfn//wfx//molden//xyz//cif/
Hint: Pressing
"5" cannot
"1" cannot
"3" cannot
"2" cannot
"0" cannot
"5" cannot
"12" cannot
"1" cannot
"2" cannot
forrtl: severe
Image
Multiwfn.exe
Multiwfn.exe
Multiwfn.exe
Multiwfn.exe
KERNEL32.DLL
ntdll.dll
C:\Multiwfn_3.8_dev_bin_Win64>move /Y density.cub density3.cub
系统找不到指定的文件。
C:\Multiwfn_3.8_dev_bin_Win64>move /
系统找不到指定的文件。
C:\Multiwfn_3.8_dev_bin_Win64>Multiwfn
Error: Unable
Multiwfn
Version
Developer: Tian
Multiwfn ///multiwfn
Multiwfn English forum: http:
Multiwfn Chinese forum: http:
( Number of parallel threads: 4 Current date: 2025-05-30 Time: 20:33:30 )
Both following papers ***MUST BE CITED IN MAIN TEXT*** if Multiwfn is used:
Tian Lu, Feiwu Chen, J. Comput. Chem., 33, 580 (2012) DOI: 10.1002/
Tian /5.0216272
See in Multiwfn binary package for more information
Now input file path, for example, E:\Planetarian\Yumemi_Hoshino.mwfn
(.wfn//wfx//molden//xyz//cif/
Hint: Pressing
"5" cannot
"1" cannot
"3" cannot
"2" cannot
"0" cannot
"5" cannot
"12" cannot
"1" cannot
"2" cannot
forrtl: severe
Image
Multiwfn.exe
Multiwfn.exe
Multiwfn.exe
Multiwfn.exe
KERNEL32.DLL
ntdll.dll
C:\Multiwfn_3.8_dev_bin_Win64>move /Y density.cub density4.cub
系统找不到指定的文件。
C:\Multiwfn_3.8_dev_bin_Win64>move /
系统找不到指定的文件。
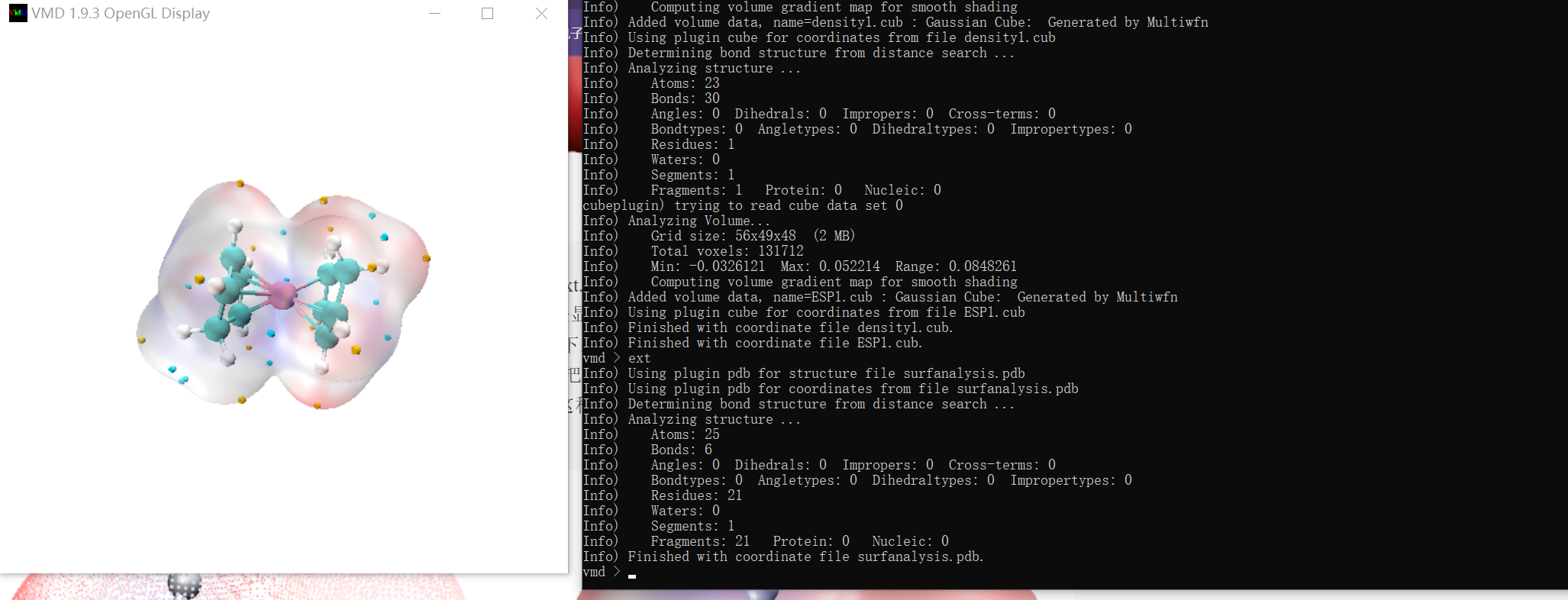
C:\Multiwfn_3.8_dev_bin_Win64>move /Y *.cub
C:\Multiwfn_3.8_dev_bin_Win64\density1.cub
C:\Multiwfn_3.8_dev_bin_Win64\ESP1.cub
移动了 2 个文件。
|